γ-氨基丁酸(GABA)是一种四碳非蛋白质氨基酸,是由谷氨酸脱羧酶不可逆、专一地将L-谷氨酸的α-羧基脱去-分子CO2得到,是高度溶于水的两性离子,pK值为4.03和10.56,因此表现出与该类氨基酸类似的生理活性,即作为一种抑制性神经递质存在于哺乳动物的中枢神经系统中。目前广泛应用于工业、动物饲料、医药和食品行业。
大量研究表明,GABA有促进睡眠、增强记忆力、抗焦虑、预防和治疗癫痫、延缓脑衰老、舒缓血管、调节激素分泌、提高受精率、解除氨毒、增强肝功能等作用。
目前制备GABA的方法主要有植物富集法、化学合成法、微生物发酵法、酶催化法。植物富集法生产GABA虽然安全环保,但GABA浓度低,尚无法用作药物、食品添加剂;化学合成法生产GABA安全性较差、有化学残留,也不易达到医药及食品行业的标准:微生物发酵法得到的GABA是一个多相复杂体系,浓度低,下游产物分离成本高,是生产高纯度、高浓度GABA的一个瓶颈问题。所以,目前制备GABA多采用酶催化法,主要是利用微生物细胞内分离得到的谷氨酸脱羧酶或能够生产谷氨酸脱羧酶的微生物细胞专一、不可逆地脱去L-谷氨酸的α-羧基,从而得到高浓度、高纯度的GABA。此方法的优点是反应条件温和、不需要昂贵的原料、能耗低,并且微生物来源的谷氨酸脱羧酶和能够生产谷氨酸脱羧酶的微生物细胞可通过简单的细胞培养大量获取。
目前大多研究以大肠杆菌为宿主表达GAD,催化生产GABA。在重组菌发酵过程中添加一定浓度的辅酶磷酸吡哆醛(PLP)能够促进GAD的折叠,从而提高GAD酶活力。除此之外,酶催化法即利用GAD将谷氨酸脱去一分子CO2制备GABA过程中,在酶转化体系中添加一定量的PLP可以促进酶液或全细胞的转化旧。有研究者报道了在大肠杆菌发酵过程中直接添加0.02mmol/LPLP后,GABA产量是对照的2.0~2.5倍。但是PLP价格昂贵,在发酵过程和酶转化体系中直接添加无疑大大增加生产成本。由于大肠杆菌自身具备吡哆醛激酶(PdxK)基因和磷酸吡哆醇氧化酶(PdxH)基因,从而形成PLP补救途径(见图1)。辅酶前体吡哆醇(PN)、吡哆胺(PM)和吡哆醛(PL)上57羟甲基通过PdxK使其磷酸化,生成对应的辅酶中问体磷酸吡哆醇(PNP)和磷酸吡哆胺(PMP)以及辅酶磷酸吡哆醛(PLP),PdxH进一步催化PNP或PMP的氧化反应,生成辅酶PLP,辅酶PLP的增加可促进GAD的折叠,提高GAD酶活力。黄燕等研究了在大肠杆菌发酵过程中添加PN后,酶活是对照组(不添加PN)的1.8倍。
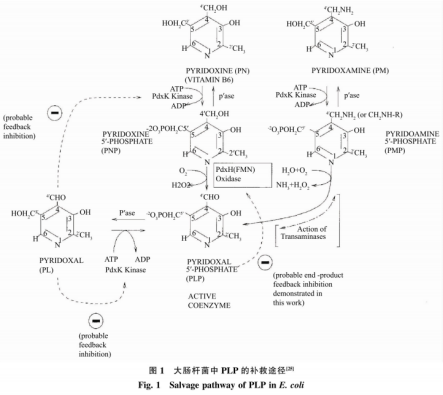
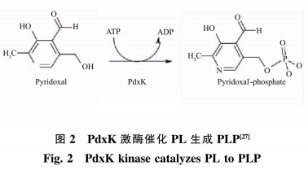
但是大肠杆菌为非食品安全菌株,在发酵过程中易产生内毒素,并且需要添加价格昂贵且有毒害作用的IPTG作为诱导剂诱导产酶,其生产得到的GAD安全性不高,距离满足食品和医药领域的要求还有一定差距。枯草芽孢杆菌是通过美国食品和药物管理局GARS认证的安全菌株,广泛应用在食品、医药生产中。然而在枯草芽孢杆菌中缺少PdxH,所以无法利用添加辅酶前体PN的方式,通过PLP补救途径将PN转化为PLP,从而提高GAD酶活力。目前,大量的研究集中在以枯草芽孢杆菌为宿主生产GAD,在酶转化过程中通过直接添加辅酶PLP提高转化效率,如丁伟等运用安全宿主枯草芽孢杆菌生产GABA,在其转化过程巾直接添加PLP,全细胞催化GABA产量为239.91g/L,转化率54.48%。由于PLP价格昂贵,导致生产成本较高,无法适应工业化生产需求。关于在枯草芽孢杆菌为宿主生产GAD发酵过程中,添加价格低廉的辅酶前体以提高GAD酶活力的研究未见报道。因此作者以枯草芽孢杆菌(Bacillussubtilis)为宿主,通过基因工程将大肠杆菌来源的谷氨酸脱羧酶基因(gadB)在B.sMbtilis中异源表达,添加相对廉价的辅酶前体PL,通过PdxK介导的PLP补救途径将其转化生成PLP(见图2),从而提高酶活和产量,降低生产成本。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒
质粒EcoliJMl09/pET-24a(+)-gadB为作者所在实验室前期构建,菌株B.5Mbtilis和表达载体pHY300PLK(含启动子PHfpaH和ParmyQ,不含信号肽)由作者所在实验室保存。
1.1.2 培养基
LB液体培养基(g/L):分别称取氯化钠10g、酵母粉5g、蛋白胨10g,溶于1L去离子水中,并通过添加2mol/LNaOH或HCl调节pH至7.0。
LB固体琼脂培养基:LB液体培养基中添加质量浓度为20g/L的琼脂粉。
高渗液体培养基:LB液体培养基巾添加0.5mol/L山梨醇。
电转培养基:LB液体培养基中分别添加0.5mol/L山梨醇和甘露醇,质量浓度为100g/L的葡萄糖。
RM培养基:LB液体培养基中添加0.5mol/L山梨醇和0.38mol/L甘露醇。
TB培养基(g/L):分别称取酵母粉24g、蛋白胨12g、甘油5g、三水磷酸氢二钾16.43g、磷酸二氢钾2.31g,溶于1L去离子水中,并添加2mol/LNaOH或HCl调节pH至7.0。
1.1.3 试剂
pH至7.0。1.1.3试剂QuickcutHindⅢ限制性内切酶:购自TaKaRa生物医药技术有限公司;2xphantaMaxMasterMix、ExnaseⅡ无缝连接试剂盒:购自诺唯赞(南京)生物科技有限公司;DL-10000DNAMarker、氨苄青霉素、四环素:购白宝生物有限公司;质粒提取试剂盒、琼脂糖凝胶DNA同收试剂盒:购白天根生化科技(北京)有限公司
UnstainedProteinMWMarker标准蛋白品、蛋白胶制备试剂盒:购自碧云天生物技术有限公司:分子级的蛋白胨和酵母粉:购自英国Oxiod公司:其他常规试剂:购自国药化学试剂有限公司。
1.1.4 主要仪器
PCR仪:购自美国Bio-Rad公司:722型紫外可见分光光度计:购白上海仪电分析仪器有限公司:冷冻式离心机:购自Beckmancoulter公司:KQ-250E型超声波细胞粉碎机:购自宁波新芝生物科技股份有限公司;pH计:购自瑞士Mettler-Toledo公司:高效液相色谱仪:购自安捷伦科技有限公司。
1.2 实验方法
1.2.1 引物设计
以质粒pET-24a(+)-gadB为模板,根据无缝克隆同源臂设计原则分别设计正、反向引物:正向弓I物(F1):57-GGAGTGTCAAGAATGGACCAGAAGCTGTTAACGGA-3':反向引物(R1):5'-‘ITTATTACCAAGCTTTCAGGTGTGTTTAAAGCTGTTC-3’。
以质粒载体pHY300PLK为模板,根据无缝克隆同源臂设计原则分别设计正、反向引物:
正向引物(F2):5'-TCAAATAAGGAGTGTCAAGAATG-3’:反向引物(R2):5’-GGTGTTTTTTTACCAAGCTT-3’。
将设计好的引物送至苏州金唯智生物科技有限公司合成。
1.2.2 目的基因和表达载体的获取
以实验室保存的质粒pET-24a(+)-gadB为模板,F1/R1为正反向引物扩增gadB目的基因;PCR反应体系(50μL):5×PSBuffer10μL、ddH2034μL、dNTP4μL、模板DNA0.5μL,正、反向引物各0.5μL,PrimerSTARPolymerase0.5μL。
PCR程序:94℃预变性5min,98℃变性10S,55℃退火5S。72℃延伸2min,变性至延伸过程进行29个循环:72℃延伸10min;4℃保存。
以质粒载体pHY300PLK为模板,F2/R2为正反向引物扩增表达载体;PCR反应体系(50μL):5×PSBuffer10μL、ddH2O34μL、dNTP4μL、模板DNA0.5μL,正、反向引物各0.5μL,PrimerSTARPolymerase0.5μL。
PCR程序:94℃预变性5min,98℃变性10s,55℃退火5S,72℃延伸6min,变性至延伸过程进行29个循环:72℃延伸10min;4℃保存。
目的基因和表达载体PCR条带通过琼脂糖核酸电泳进行验证。
1.2.3 目的基因和表达载体的连接与鉴定
1.2.2已验证正确的PCR产物通过琼脂糖凝胶同收试剂盒进行回收。回收后的PCR产物用ExnaseⅡ连接酶连接,其连接体系如下:ddH2O5.3μL、5×CEBuffer2μL、ExnaseⅡ1μL、表达载体1.16μL、目的基因0.54μL,置于PCR仪37℃反应30min完成连接。
采用大肠杆菌JMl09热击转化法后,将转化细胞液涂布至含100μg/mL氨苄青霉素的LB固体培养基上,在37℃恒温培养箱倒置培养10~12h,挑取单菌落至含100μg/mL氨苄青霉素的10mLLB液体培养基中,置于恒温摇床37℃、200r/min培养8~10h后收集菌体,并提取质粒以便验证和进行后续实验。将提取的质粒进行HindⅢ酶切验证正确后送往苏州金唯智生物科技有限公司测序。将测序正确的质粒通过电击转化法导入表达宿主B.Subtilis中,将转化细胞液涂布至含20μg/mL四环素的LB同体培养基上,在37℃恒温培养箱倒置培养10~12h,挑取单菌落至含20μg/mL四环素的10mLLB液体培养基中,置于恒温摇床37℃、200r/min培养8~10h后收集菌体,并提取质粒进行酶切验证。
文章版权备注
- 2023-05-01奶味香精的制备技术与开发现状
- 2023-04-04阿魏酸及其衍生物在食品添加剂领域研究进展(一)
- 2023-03-23甘肃省榆中县市场监督管理局强化食品快检 筑牢食品安全防线
- 2023-03-23常德:专项整治酒类市场12种违法行为
- 2023-03-23贵州这7批次食品检出食品添加剂问题,有食用植物调和油、冰糖大蒜、无油剁椒等
- 2023-03-23怀化:部署开展制止餐饮浪费专项行动
- 2023-03-23超90%展商已确认展位 6月食品原料展带来海内外商机
- 2023-03-23关于召开2023年国际食品安全与健康大会的通知
- 2023-03-23河南省市场监管局召开落实食品安全“两个责任”暨制止餐饮浪费专项行动调度视频会议
- 2023-03-23邵阳市食品安全“两个责任”机制推进暨野生蘑菇中毒防控部署电视电话会议召开
 豫ICP备19024296号
豫ICP备19024296号 售前咨询
售前咨询