谷氨酸脱羧酶在枯草芽孢杆菌中的表达及应用(二)
1.2.4 摇瓶发酵生产CAD及蛋白质检测
将-80℃保存的重组菌B.subtilis/pHY300PLK-gadB以体积分数0.2%接种至含20μg/mL四环素的LB液体培养基中,恒温摇床37℃、200r/min扩大培养8~10h,再以体积分数5%转接至含20μg/mL四环素的TB发酵培养基中,恒温摇床37℃、200r/min培养2~3h后分别加入一定浓度的PLP、PL、PN,于恒温摇床33℃、200r/min进行重组蛋白质的表达。发酵过程中在不同时间取样,测0D600。12000r/min离心1min得到菌体和发酵上清液。将菌体用1mL、50mmol/L、pH5.0的Na2HP04-柠檬酸缓冲液重悬,加入0.6mg/mL溶菌酶,在37℃反应30min后置于冰水中超声破碎,12000r/min离心5min得到破壁上清液和破壁沉淀,使用SDS-PAGE检测其蛋白质。
1.2.5 重组CAD酶活测定底
物溶液配制:0.1mol/L-水谷氨酸钠和0.15mmol/LPLP溶于50mmol/L、pH4.5的Na2HPO4-柠檬酸缓冲液中,置于4℃避光保存。
反应体系:将装有360uL底物溶液的1.5mLEP管于37℃水浴锅预热10min后,加入40uLGAD粗酶液,在37℃下反应4min后,加入600uL、0.2mol/L、pH10的硼酸缓冲液终止反应,置于沸水中灭活10min。
GABA生成量测定:采用HPLC-OPA氨基酸柱前衍生法检测GABA生成量。
酶活定义:在反应液中,1min催化底物转化生成1umolGABA所需的酶量为一个活力单位(U)。
本文中重组GAD均指重组菌所产的GAD。
1.2.6 重组菌全细胞制备GABA工艺流程
1) GABA的制备
水配制质量浓度为127g/L的一水谷氨酸钠底物(折算成100g/L谷氨酸,以下均以折算后的谷氨酸质量浓度进行阐述)。在150mL三角锥形瓶中加入20mL底物,加入一定量的湿菌体(在55℃水浴锅预处理50min,改善细胞膜的通透性),反应过程中,每隔2h用0.6mol/L的H2SO4和NaOH调节pH,使其始终与初始pH一致。在一定温度、pH下,150r/min水浴摇床中反应一段时间,终止反应后进行煮沸处理,12000r/min离心5min得上清液,适当稀释并过0.22μm有机滤膜后用HPLC-OPA柱前衍生法进行检测。
2) GABA生成量的测定
将过滤好的样品通过HPLC检测GABA生成量。HPLC色谱条件如下:Agilent1200HPLC色谱仪、Agilent自动进样器、GLInertsilODS-3液相柱、Agilent紫外检测器。流动相A:准确量取995mL纯净水,加入4.52g无水乙酸钠,搅拌使其充分溶解,再加入5mL四氢呋喃和0.2mL三乙胺,之后用冰乙酸调节pH至7.20±0.05,充分混合后用0.22μm无机纤维素滤膜过滤备用:流动相B:准确量取200mL纯净水,加入4.52g无水乙酸钠,搅拌使其充分溶解,用冰乙酸调节pH至7.20±0.05后,再依次加入400mL色谱纯的乙腈和甲醇,用冰乙酸调节pH至7.20+0.05,混合后用0.22μm有机尼龙滤膜过滤备用。梯度洗脱,流量为0.8mL/min,柱温为40℃。根据吸收峰面积和GABA标准品峰面积计算GABA生成量,计算公式如下:
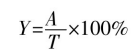
式中:Y为GABA转化率,%;4为谷氨酸转化生成GABA的实际质量浓度,g/L;T为谷氨酸转化生成GABA的理论质量浓度,g/L。
2 结果与讨论
2.1 产谷氨酸脱羧酶重组菌的构建
以实验室保存的质粒pET-24a(+)-gadB为模板,扩增gadB目的片段,并以质粒载体pHY300PLK为模板,扩增表达载体片段。扩增得到的产物用琼脂糖凝胶电泳检测,目的基因片段gadB和质粒表达载体pHY300PLK长度约为1428bp和5731bp,与理论碱基大小一致。验证正确后通过ExnaseⅡ连接酶将两个基因片段连接再转入E.coliJMl09感受态细胞,涂布到LB固体培养基(含氨苄青霉素抗性)后挑取单菌落至LB液体培养基(含氨苄青霉素抗性)中过夜培养。提取质粒,酶切验证和测序成功后,将重组质粒用电击转化法导入表达宿主B.subtilis并涂布到LB固体培养基(含四环素抗性),之后挑取单菌落至LB液体培养基(含四环素抗性)中培养8~10h,提取质粒后进行酶切验证分析。结果见图3,分别在5130bp和2100bp处有明显的条带,说明重组表达质粒pHY300PLK-gadB在B.subtilis中构建成功。
2.2 分别添加辅酶及辅酶前体对重组菌摇瓶发酵的影响
2.2.1 添加不同种类辅酶及辅酶前体对重组菌产酶情况的影响
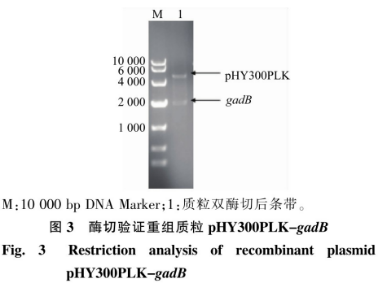
种类辅酶及辅酶前体对重组菌产酶情况的影响重组菌按照1.2.4进行摇瓶发酵产酶,摇瓶发酵温度33℃,在重组菌发酵过程中分别添加辅酶PLP(简称GAD-PLP)、辅酶前体PL(简称GAD-PL)和PN(简称GAD-PN)至终浓度为0.5mmol/L。发酵结束后12000r/min离心1min获得菌体,用超声波细胞粉碎机进行细胞破碎,12000r/min离心5min得到GAD粗酶液。结果见图4,诱导48h后,酶活分别达到25.40、28.14U/mL和15.55U/mL,是对照GAD-0酶活(16.34U/mL)的1.55、1.72和0.95倍。由此可知,添加0.5mmol/LPN时,酶活相较于对照并无明显区别,这是因为枯草芽孢杆菌中缺少PdxH氧化酶,无法通过PLP补救途径将PN转化为PLP,从而提高GAD酶活力。其次随着诱导培养时间的延长,细胞内合成的少量PLP被自身吸收并消耗而无法满足GAD表达水平提高的需要。然而向培养基巾添加适量的PLP或PL可以直接或问接通过PLP补救途径合成PLP,从而提供能够维持GAD稳定的辅酶PLP,促进GAD的折叠,提高酶活。鉴于PLP成本高于PL,所以在发酵过程中选择添加PL进行后续实验。
2.2.2 不同吡哆醛浓度对重组菌产酶情况的影响
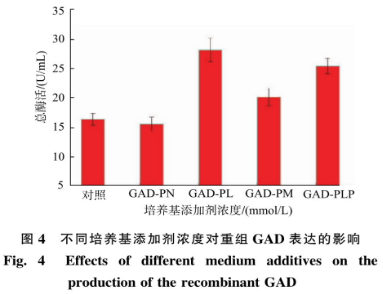
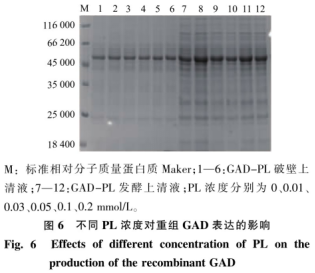
重组菌按照1.2.4进行摇瓶发酵,在过程中添加不同浓度的PL(0.01、0.03、0.05、0.07、0.1、0.2、0.3mmol/L)发酵培养48h后,12000r/min离心1min获得菌体,超声波细胞破碎机进行细胞破碎,12000r/min离心5min得到GAD粗酶液,结果见图5。随着PL浓度的增加,GAD酶活也随之提高,当PL浓度为0.1mmol/L时,GAD酶活达到最高为28.28U/mL。继续增加PL的浓度,酶活并未发生显著变化,故可知PL的最适添加浓度为0.1mmol/L。图6为发酵过程中添加不同浓度PL后的重组GAD蛋白质电泳图,从图中可见在相对分子质量53000的条带附近有一条清晰的蛋白质电泳条带,符合GAD理论蛋白质相对分子质量大小。
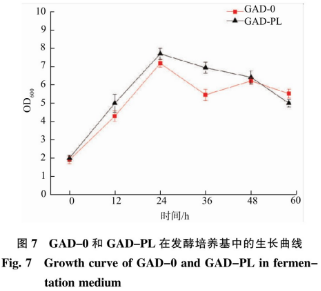
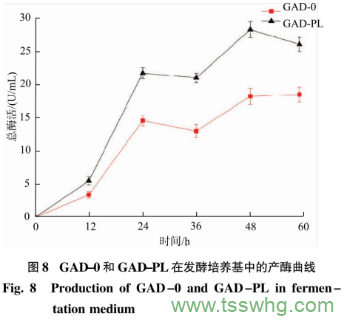
2.2.3 添加吡哆醛前后重组菌发酵过程比较
重组菌按照1.2.4进行摇瓶发酵,在过程中添加0.1mmol/LPL,探究其在不同发酵时间(0、12、24、36、48、60h)对重组菌生长(见图7)和产GAD情况(见图8)的影响。与GAD-0相比,在发酵时问36h左右,PL对菌体的生长有一定的促进作州,是因为存蛋白质表达过程中,通过PLP补救途径,PdxK激酶将PL磷酸化形成PLP,促进GAD的折叠,一定程度上有利于菌体生长。随着发酵时问的延长,南于PLP稳定性差而失去作用,导致菌体的生长有所下降。由图8可知,与GAD-0相比,GAD-PL酶活最高达到28.28U/mL,添加适量的PL可以提供给GAD未知浓度的PLP,从而提高GAD酶活。
声明:本文所用图片、文字来源《食品与生物技术》,版权归原作者所有。如涉及作品内容、版权等问题,请与本网联系
相关链接:谷氨酸钠,氧化酶,琼脂糖凝胶
文章版权备注
- 2023-05-01不同贮藏方式对红香酥梨采后生理及品质的影响(一)
- 2023-05-01奶味香精的制备技术与开发现状
- 2023-05-01双孢蘑菇液体菌种发酵及栽培效果浅析
- 2023-05-01菌种强化结合工艺优化提高酱香白酒基酒中四甲基吡嗪含量的研究(二)
- 2023-05-01发酵小麦胚芽产2,6-二甲氧基对苯醌菌种筛选及发酵条件优化(一)
- 2023-05-01猕猴桃中铁含量的测定与测定因素的探究
- 2023-05-01腐植酸调节砷酸盐生菜毒性作用研究(四)
- 2023-05-01腐植酸调节砷酸盐生菜毒性作用研究(三)
- 2023-05-01腐植酸调节砷酸盐生菜毒性作用研究(二)
- 2023-05-01腐植酸调节砷酸盐生菜毒性作用研究(一)
 豫ICP备19024296号
豫ICP备19024296号 售前咨询
售前咨询